National Technical University of Athens School of Chemical Engineering Department of Materials Science and Engineering Computational Materials Science and Engineering Group (Co.M.S.E.) |
| News 17 March 2026 DTU invites applications for an Associate or Tenure Track Assistant Professor position in Applied Thermodynamics. Click here for the more details. 17 March 2026 Sincerest congratulations to our former group member, Dr. Georgios G. Vogiatzis, on his appointment as Assistant Professor in the Department of Chemical Engineering at the University of Patras. We warmly congratulate him on this achievement and wish him continued success in his academic career. 17 March 2026 Sincerest congratulations to our former group member, Dr. Aristotelis P. Sgouros, on his appointment as Associate Researcher at the Theoretical and Physical Chemistry Institute of the National Hellenic Research Foundation. We warmly congratulate him on this achievement and wish him continued success in his research career. 24 January 2026 Ph.D. thesis presentation: On the 3rd of February 2026, Georgios Mikaelian will present his Ph.D. Thesis, entitled "In-Silico Study of Fluoxetine, Dopamine, Berubicin, and Doxorubicin in Biomolecular and Drug Delivery Systems" in front of his seven-member examination committee. The presentation will take place in the "Nikos Koumoutsos" hall of the Chemical Engineering building at 12:00. Click here for the more details. 2 December 2025 We are delighted to announce the release of Multiscale Modelling of Polymers by D. N. Theodorou and V. G. Mavrantzas, published in the Oxford Series on Materials Modelling. Click here for the more details. 1 December 2025 The Online Training Workshop on Computational Materials Science 2025 (CMS25) will take place on 13-14 December via ZOOM. Organized by the National Hellenic Research Foundation under the auspices of EEETSY. Click here for the more details. 10 November 2025 A PhD program titled "Molecular Modeling of Sustainable Heat Batteries (RecyWax+)" is available in the group of Profs. Alexey Lyulin, Heiner Friedrich, and Henk Huinink (TU/e), in collaboration with Prof. Bernard Geurts (University of Twente). Click here for the more details. 30 June 2025 A PhD program with title "PhD Program in Process and Product Engineering" is available in the group of Profs. Giovanni Ianniruberto and Pino Marrucci at the Universita di Napoli. Click here for the more details. Click here for the more details. 12 June 2025 The European Materials Informatics Network (EUMINe) invites students/scientists in the field of materials science to participate in the upcoming EUMINe Training School which will take place on 19-20 June 2025 at the Alex Castella Hotel in Piraeus, Greece. Click here for the more details. 31 March 2025 We would like to invite you to our group's next meeting, which will take place next Tuesday, April 1st, at 14:00, in the "N. Koumoutsou" room of the School of Chemical Engineering, NTUA. Professor Theodoros Karakasidis, Chairman of the Department of Physics at the University of Thessaly, will discuss an extremely interesting and timely topic, namely the prediction of material properties using machine learning methods. Click here for the abstract of the presentation. 9 May 2024 A summer school/Workshop is organized by the Department of Mathematics of the University of the Aegean, in cooperation with the Hellenic Society of Rheology (HSR). Τhe event is open to all M.Sc. and Ph.D. students, post-docs, scientists and engineers conducting research or interested in the field of rheology and fluid mechanics. The program will consist of 6 short courses, invited lectures, a poster session and a limited number of oral presentations. All topics in theoretical, computational, and experimental rheology are welcome. Click here for more information. 28 March 2024 The Chemical Engineering department of the Technical University of Denmark (DTU) is seeking a motivated PhD student who is willing to be part of a world-leading research environment and contribute to the development of thermodynamic models and algorithms for electrolyte solutions. The appointment of this PhD position will be at the Center for Energy Resources Engineering (CERE), Department of Chemical and Biochemical Engineering. Click here for more information. PhD and Postdoctoral opportunities in Applied Knot Theory at ASU. Click here for more information. 6 December 2023 PhD and Postdoctoral opportunities in Applied Knot Theory at ASU. Click here for more information. 20 November 2023 Two PhD positions are available within the framework of the European Network "ReBond". Click here and here for more information. 19 September 2023 PhD position available in Computational Chemistry/Polymers Degradation funded by PlasticUnderground Marie Curie ITN. Click here for more information. 2 September 2023 Masters, PhD, Postdoctoral and Project-Manager positions are available at FORTH-IESL in the field of Synthesis, Experiments and Modelling/Simulations of Soft Matter systems (with emphasis in Polymers and Colloids) for Green and sustainable applications. Click here for more information. 2 September 2023 A PhD position on molecular-dynamics simulations of thermomechanical behaviour of epoxy-copper interfaces is available starting from January 2024 in the Soft Matter and Biological Physics (SMB) group of the Applied Physics department of TU/e. The project will be supervised by prof. dr. Alexey Lyulin. Click here for more information. 12 June 2023 On Friday, the 7th of July, 2023, two distinguished professors from the USA will visit the Computational Materials Science and Engineering (CoMSE) group at NTUA: Professor Bradley Chmelka (Department of Chemical Engineering, University of California, Santa Barbara) and Professor Eleni Panagiotou (School of Mathematical and Statistical Sciences, Arizona State University). In the context of their visit they will give lectures on their research work, with the following titles: Prof. Bradley Chmelka; Compositional and structural order at inorganic-organic interfaces. Prof. Eleni Panagiotou; Novel topological metrics of entanglement in polymers. The lectures will be given in the Koumoutsos room of the School of Chemical Engineering at NTU Athens, starting at 14:00. Click here for more information. 25 April 2023 The 2023 European Conference on Computational and Theoretical Chemistry, co-organized by the Greek Chemists Association and the Division of Computational and Theoretical Chemistry of the European Chemical Society will be held in Thessaloniki, 27-31 August 2023. Click here for more information. Read all news... |
Discotic liquid crystals Discotic polyaromatic molecules with flexible chains grafted around their periphery constitute an interesting class of material for organic electronics applications[REF]. These molecules tend to self-organize into columnar arrangements, with the π-π interaction along the columnar direction being responsible for charge transfer phenomena and the entropic behavior of the side chains rendering the materials soluble, thus allowing greater processability in comparison to their solid-state ungrafted counterparts. Our studies focus on - but are not limited to - to a series of planar polyaromatic cores of hexagonal, trigonal and tetragonal symmetry such as the hexa-peri-hexabenzocoronene (HBC) C42H18[REF], the superphenalene C96H30 and the C132H48 molecules. In order to investigate the soft, liquid-crystalline phases of molecular crystals made of the aforementioned mesogens, a plethora of peripheral side chains is examined, such as linear or branched aliphatic chains comprised of purely sp3 carbon atoms, complex chains containing phenyl and ether groups, and semi- or fully-fluorinated aliphatic side chains. 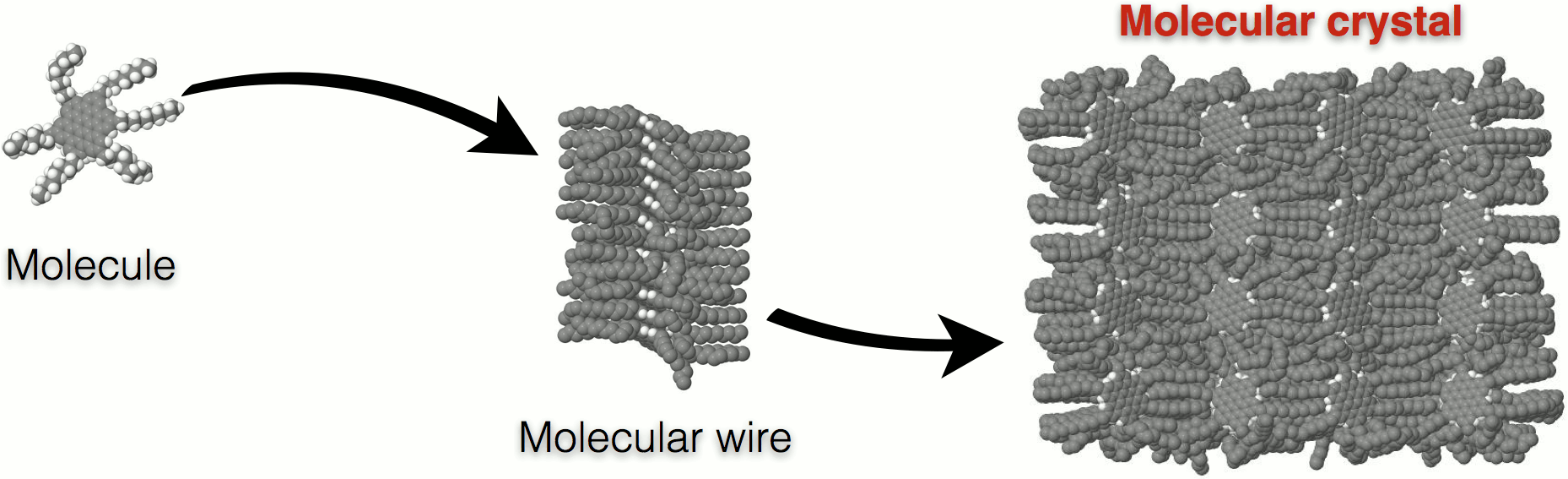 Figure 1: A HBC-C12H25 molecule forming neatly stacked molecular wires that aggregate to periodic molecular crystals. Molecular dynamics simulations are employed in order to investigate structural, thermodynamic, mechanical and dynamical properties of molecular crystals comprised of various discotic mesogens. Calculations are carried out with the LAMMPS[REF] package in the canonical, isothermal-isobaric and isothermal-isostress statistical ensembles via the application of suitable thermostat and barostat algorithms in order to equilibrate initial configurations and accumulate trajectories over sufficient time intervals for the determination of various properties. Intramolecular and intermolecular interactions are modeled by means of empirical force fields taken from the literature[REF][REF][REF][REF]. Bonded interactions are quantified utilizing harmonic bond and angle and proper dihedral angle terms. Non-bonded interactions, namely the electrostatic and van der Waals interactions, are modeled through the Coulomb and Lennard-Jones potentials respectively, with the partial charges needed for the Coulomb potential extracted from Mulliken population analyses based on a semi-empirical tight-binding Hamiltonian using the MOPAC package[REF]. All systems are examined in their bulk phase, which is approximated using full periodic boundary conditions and appropriate mesh-based techniques and/or tail corrections associated with non-bonded interaction calculations. 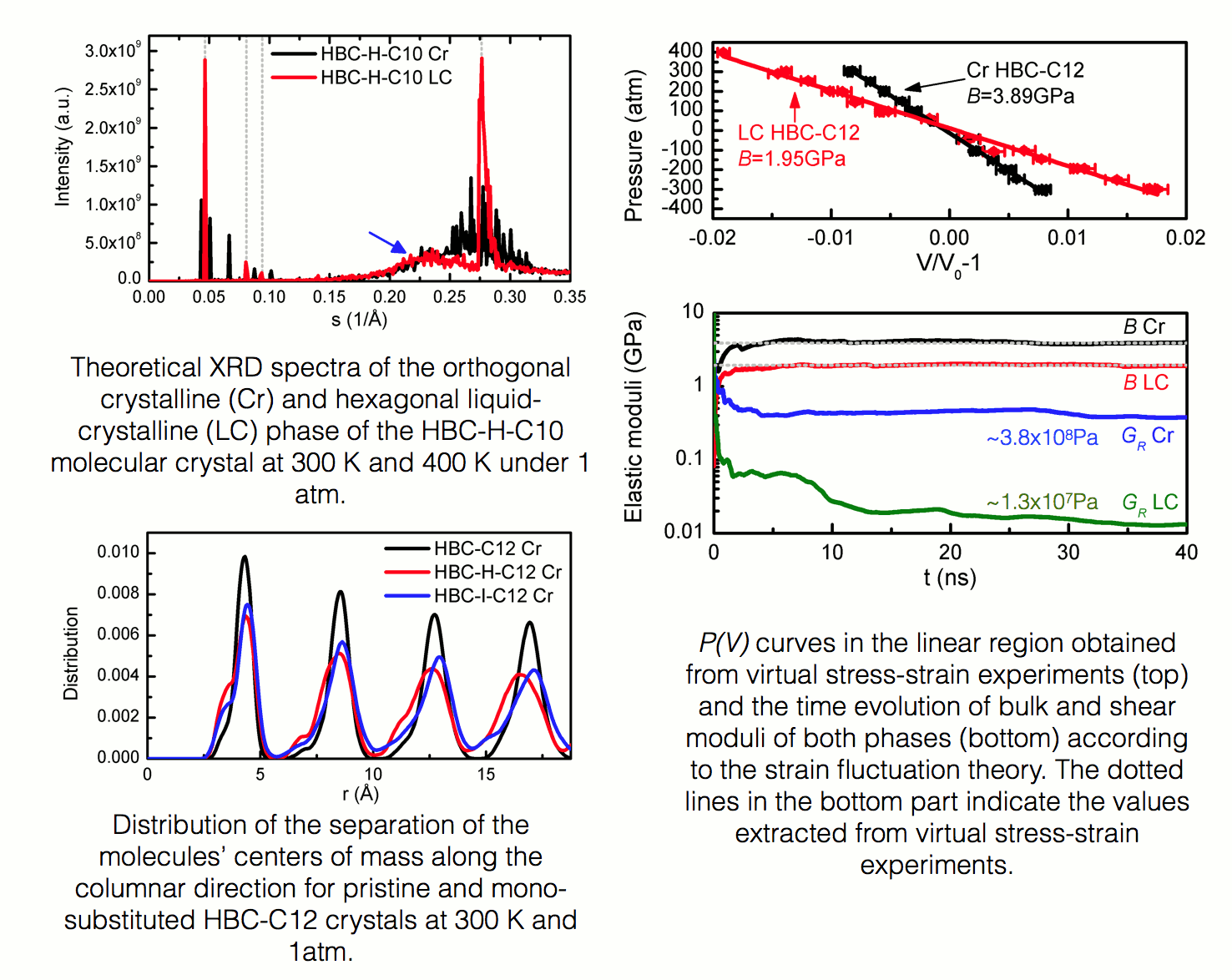 Figure 2: Structural and mechanical characterization of various HBC-based systems in the crystalline (Cr) and liquid crystalline (LC) phases. The major scope of our analyses is to examine the effect of the chemical composition and geometry of the basis molecule on the properties of the molecular crystals at given temperature and pressure conditions. A detailed knowledge of the relationships between composition of the discotic molecule, phase behavior, and structural and dynamical properties of the condensed phases formed is crucial for the manufacture of efficient materials for organic electronic applications. This is because structural characteristics, such as the core-to-core distance and stacking defects, alongside with temporal phenomena, such as intracolumnar molecular displacements, vibrations and collective columnar motions can alter the electronic properties of the systems dramatically. Structural and dynamical properties calculations are complemented by thermodynamic and mechanical theoretical characterization, leading to a full survey of material properties for every mesogen, providing insight for intelligent materials design. Relevant publications
Relevant projects
|
|
 |
 |
|

| Computational Materials Science and Engineering Group Department of Materials Science and Engineering School of Chemical Engineering, National Technical University 9 Heroon Polytechniou Street, Zografou Campus, 15780, Athens, Greece Tel. +30 210 772 3216, Fax +30 210 772 3112 Webmaster: comse.chemeng.ntua@gmail.com |
Copyright © 2023 - All rights reserved |