National Technical University of Athens School of Chemical Engineering Department of Materials Science and Engineering Computational Materials Science and Engineering Group (Co.M.S.E.) |
| News 17 March 2026 DTU invites applications for an Associate or Tenure Track Assistant Professor position in Applied Thermodynamics. Click here for the more details. 17 March 2026 Sincerest congratulations to our former group member, Dr. Georgios G. Vogiatzis, on his appointment as Assistant Professor in the Department of Chemical Engineering at the University of Patras. We warmly congratulate him on this achievement and wish him continued success in his academic career. 17 March 2026 Sincerest congratulations to our former group member, Dr. Aristotelis P. Sgouros, on his appointment as Associate Researcher at the Theoretical and Physical Chemistry Institute of the National Hellenic Research Foundation. We warmly congratulate him on this achievement and wish him continued success in his research career. 24 January 2026 Ph.D. thesis presentation: On the 3rd of February 2026, Georgios Mikaelian will present his Ph.D. Thesis, entitled "In-Silico Study of Fluoxetine, Dopamine, Berubicin, and Doxorubicin in Biomolecular and Drug Delivery Systems" in front of his seven-member examination committee. The presentation will take place in the "Nikos Koumoutsos" hall of the Chemical Engineering building at 12:00. Click here for the more details. 2 December 2025 We are delighted to announce the release of Multiscale Modelling of Polymers by D. N. Theodorou and V. G. Mavrantzas, published in the Oxford Series on Materials Modelling. Click here for the more details. 1 December 2025 The Online Training Workshop on Computational Materials Science 2025 (CMS25) will take place on 13-14 December via ZOOM. Organized by the National Hellenic Research Foundation under the auspices of EEETSY. Click here for the more details. 10 November 2025 A PhD program titled "Molecular Modeling of Sustainable Heat Batteries (RecyWax+)" is available in the group of Profs. Alexey Lyulin, Heiner Friedrich, and Henk Huinink (TU/e), in collaboration with Prof. Bernard Geurts (University of Twente). Click here for the more details. 30 June 2025 A PhD program with title "PhD Program in Process and Product Engineering" is available in the group of Profs. Giovanni Ianniruberto and Pino Marrucci at the Universita di Napoli. Click here for the more details. Click here for the more details. 12 June 2025 The European Materials Informatics Network (EUMINe) invites students/scientists in the field of materials science to participate in the upcoming EUMINe Training School which will take place on 19-20 June 2025 at the Alex Castella Hotel in Piraeus, Greece. Click here for the more details. 31 March 2025 We would like to invite you to our group's next meeting, which will take place next Tuesday, April 1st, at 14:00, in the "N. Koumoutsou" room of the School of Chemical Engineering, NTUA. Professor Theodoros Karakasidis, Chairman of the Department of Physics at the University of Thessaly, will discuss an extremely interesting and timely topic, namely the prediction of material properties using machine learning methods. Click here for the abstract of the presentation. 9 May 2024 A summer school/Workshop is organized by the Department of Mathematics of the University of the Aegean, in cooperation with the Hellenic Society of Rheology (HSR). Τhe event is open to all M.Sc. and Ph.D. students, post-docs, scientists and engineers conducting research or interested in the field of rheology and fluid mechanics. The program will consist of 6 short courses, invited lectures, a poster session and a limited number of oral presentations. All topics in theoretical, computational, and experimental rheology are welcome. Click here for more information. 28 March 2024 The Chemical Engineering department of the Technical University of Denmark (DTU) is seeking a motivated PhD student who is willing to be part of a world-leading research environment and contribute to the development of thermodynamic models and algorithms for electrolyte solutions. The appointment of this PhD position will be at the Center for Energy Resources Engineering (CERE), Department of Chemical and Biochemical Engineering. Click here for more information. PhD and Postdoctoral opportunities in Applied Knot Theory at ASU. Click here for more information. 6 December 2023 PhD and Postdoctoral opportunities in Applied Knot Theory at ASU. Click here for more information. 20 November 2023 Two PhD positions are available within the framework of the European Network "ReBond". Click here and here for more information. 19 September 2023 PhD position available in Computational Chemistry/Polymers Degradation funded by PlasticUnderground Marie Curie ITN. Click here for more information. 2 September 2023 Masters, PhD, Postdoctoral and Project-Manager positions are available at FORTH-IESL in the field of Synthesis, Experiments and Modelling/Simulations of Soft Matter systems (with emphasis in Polymers and Colloids) for Green and sustainable applications. Click here for more information. 2 September 2023 A PhD position on molecular-dynamics simulations of thermomechanical behaviour of epoxy-copper interfaces is available starting from January 2024 in the Soft Matter and Biological Physics (SMB) group of the Applied Physics department of TU/e. The project will be supervised by prof. dr. Alexey Lyulin. Click here for more information. 12 June 2023 On Friday, the 7th of July, 2023, two distinguished professors from the USA will visit the Computational Materials Science and Engineering (CoMSE) group at NTUA: Professor Bradley Chmelka (Department of Chemical Engineering, University of California, Santa Barbara) and Professor Eleni Panagiotou (School of Mathematical and Statistical Sciences, Arizona State University). In the context of their visit they will give lectures on their research work, with the following titles: Prof. Bradley Chmelka; Compositional and structural order at inorganic-organic interfaces. Prof. Eleni Panagiotou; Novel topological metrics of entanglement in polymers. The lectures will be given in the Koumoutsos room of the School of Chemical Engineering at NTU Athens, starting at 14:00. Click here for more information. 25 April 2023 The 2023 European Conference on Computational and Theoretical Chemistry, co-organized by the Greek Chemists Association and the Division of Computational and Theoretical Chemistry of the European Chemical Society will be held in Thessaloniki, 27-31 August 2023. Click here for more information. Read all news... |
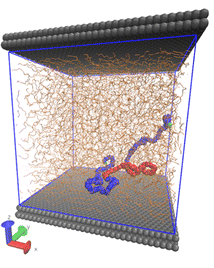
Polymer melts at interfaces Understanding the fascinating and complex dynamics of melts of large flexible polymer coils close to solid substrates has been an ongoing challenge for many decades. From the point of view of molecular simulations, the spectrum of length and time scales associated with polymer melts of long chains poses a formidable challenge to studying the long-time dynamics. We have formulated a method, based on combining self-consistent field theory with dynamically corrected transition state theory, for estimating the rates of adsorption and desorption of end-constrained chains e.g., by cross-links or entanglements) from a polymer melt onto a solid substrate. This approach has been successfully applied on a polyethylene/graphite system, where the whole methodology was parameterized by atomistically detailed molecular simulations. For short-chain melts, which can still be addressed by molecular dynamics simulations with reasonable computational resources, the self-consistent field approach gives predictions of the adsorption and desorption rate constants which are gratifyingly close to molecular dynamics estimates.  Figure 1: Schematic illustration of an end-constrained subchain of 50 CH2 units (marked in red), whose residence times in the adsorbed and desorbed state, in the course of an MD simulation, were used for estimating adsorption and desorption kinetics via hazard-plot analysis. External link: http://pubs.acs.org/doi/abs/10.1021/ma501454t To the best of our knowledge, this the first time that the solution of the polymer SCF problem next to a solid surface has been used to provide insight into the dynamics of the system. This work suggests that SCF, coupled with transition-state theory, can give good results for the rate of adsorption/ desorption between polymers in a melt and a surface. SCF theory is less computationally demanding than atomistic simulations, and thus SCF can be used to examine regimes not practically accessible to atomistic simulations, such as the regimes of low adsorption or low desorption rates. Furthermore, the parameters in SCF have been directly connected to atomistic parameters, suggesting that SCF may provide a more faithful description of the problem than alternative, coarse-grained simulation methods. In the regime where molecular dynamics simulation of adsorption rates is practical (short chains, very close to the surface), our results for the adsorption rate are in good agreement with the results from molecular dynamics simulation. Finally, a reasonable agreement between the melt adhesion tension computed from SCF and that measured in atomistic simulations and in experiment, was found. 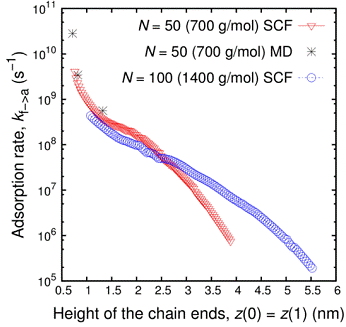 Figure 2: Adsorption rate constant, predicted by Self Consistent Field / Transition State Theory calculations, is presented as a function of the distance of the ends of an end-constrained subchain from the graphite surface. Both ends are kept at the same distance from the graphite surface, and we consider chain lengths of N = 50 and N = 100 methylenes. Adsorption rates for an end-constrained C50 subchain in a C200 polyethylene melt next to graphite obtained by MD simulation using a detailed united-atom model are also shown. External link: http://pubs.acs.org/doi/abs/10.1021/ma501454t Relevant publications
Relevant projects
|
|
 |
 |
|

| Computational Materials Science and Engineering Group Department of Materials Science and Engineering School of Chemical Engineering, National Technical University 9 Heroon Polytechniou Street, Zografou Campus, 15780, Athens, Greece Tel. +30 210 772 3216, Fax +30 210 772 3112 Webmaster: comse.chemeng.ntua@gmail.com |
Copyright © 2023 - All rights reserved |